
SOFT TISSUE SARCOMA
Symptoms | Signs | Treatment
What are Soft tissue sarcomas (STS) ?
- These are heterogeneous groups of cancers arising from mesenchymal cells of the body (other than the bone and cartilage).
- They are divided and named based on tissue or cell of origin.
- Due to advancements in molecular genetics and cellular differentiation soft tissue sarcomas are being reclassified regularly based on immunohistochemistry.
What sites in body are affected ?
- STS can develop from cells of mesenchymal origin such as fat, muscle, nerves, fibrous tissues, blood vessels.
- They can arise in any part of the body, but have a tendency to develop in extremities (nearly 60 %).
- Other sites are trunk, head and neck area, internal organs, retroperitoneal cavity.
What are the causes for STS?
- Currently, most STS are believed to arise spontaneously without any specific cause.
- Few risk factors postulated are as follows:
- Radiation exposure (7 -10 years after exposure)
- Exposure to certain toxic chemicals
- Lymphedema
- Hereditary cancer syndromes
- Li-Fraumeni syndrome: Risk of radiation induced sarcoma
- Neurofibromatosis: Malignant peripheral nerve sheath tumour (MPNST)
- Gardner syndrome: Aggressive fibromatosis
- Werner syndrome: Soft tissue sarcomas
- Gorlin syndrome: Fibrosarcoma & Rhabdomyosarcoma
- Tuberous Sclerosis: Rhabdomyosarcoma
- The individuals who have any of the above mentioned disorders or risk factors are closely monitored and followed up regularly to allow early and rapid identifications of STS.
What are the types of STS?
STS are classified based on cell of origin and recently based on cytogenetic findings.
| Cell of Origin | Benign | Intermediate | Malignant |
| Adipose | Lipoma, Angiolipoma | Well differentiated liposarcoma/ atypical lipomatous tumour | Dedifferentiated, myxoid and pleomorphic Liposarcoma, |
| Fibroblastic/Myoblastic | Myositis ossificans, Elastofibroma, | Desmoid/aggressive fibromatosis, plantar fibromatosis, Solitary fibrous tumour, Dermatofibrosarcoma protuberans | Adult fibrosarcoma, Myxofibrosarcoma, Sclerosing epithelioid fibrosarcoma |
| Fibrohistiocytic | Tenosynovial giant cell tumour | Plexiform fibrohistiocytic tumour | |
| Smooth muscle | Leiomyoma, Glomus tumour | Leiomyosarcoma | |
| Skeletal muscle | Rhabdomyoma | Rhabdomyosarcoma family of tumours | |
| Vascular | Haemangioma | Kaposi sarcoma | Angiosarcoma, Epithelioid hemangioendothelioma |
| Nerve sheath | Schwannoma, Neurofibroma | Malignant peripheral nerve sheath tumour (MPNST) | |
| Uncertain Differentiation | Fibromyxoma, myxoma | Atypical fibroxanthoma, myoepithelioma | Synovial sarcoma, Epithelioid sarcoma, Alveolar soft-part sarcoma, Clear cell sarcoma, Myxoid chondrosarcoma |
| Unclassified Undifferentiated | Undifferentiated pleomorphic sarcoma (UPH), round cell, spindle cell and epithelioid variety |
How are Soft Tissue Sarcoma identified?
https://bonecancer.in/2020/05/01/arriving-at-diagnosis-services/
https://bonecancer.in/2020/05/01/biopsy/
- An individual with suspected STS presents with a Painless Lump which has Grown over a period of weeks to months.
- Rarely, if lesions are superficial the skin may breakdown leading to ulcers and fungation.
- Lesions in the pelvic or retroperitoneal area often present late due to the space and propensity to grow large until symptoms appear.
- Pain is an uncommon presentation.
- Loss of weight or appetite are seen in advanced cases.
Any lump more than 5 cm (size of a golf ball), growing lately or found deep to skin should be evaluated.
How are Soft Tissue Sarcoma investigated?
- Clinical examination of involved part and associated lymph nodes (to check local spread)
- Blood tests
- X-ray of Involved region & Chest
- Ultrasound examination will provide clues regarding the nature of lesions.
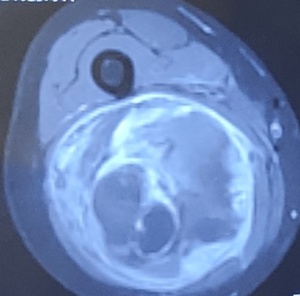
- MRI with Contrast scan is the preferred imaging investigation of choice to diagnose STS.
- MRI provides details regarding the site, size, extent, aggressiveness of sarcoma and often the origin of STS.
- These details help the treating musculoskeletal oncosurgeon to biopsy from the appropriate site of STS.CT scan, preferably with contrast, has limited value in diagnosing STS due to limited advantages when compared to MRI and the risk of extensive radiation exposure.
- The main indication is for CT Thorax (1mm slice) to pick up any metastases to lungs and complete the staging process.
- In certain situations, CT Thorax with Abdomen-pelvis is performed in absence of PET scan.
- The Core Needle/Trucut biopsy or sometimes Open Biopsy is preferred over Fnac and provides adequate tissue material to the pathologist to provide diagnosis.
- Image guided biopsy (USG or CT guided) can also be performed in certain situations.
- Certain situations may require additional Immunohistochemistry markers, Cytogenetic, FISH or RT-PCR tests to be performed to provide accurate diagnosis.
- This investigation cascade is completed by performing FDG PET CT scan to assess spread of STS in other areas of the body.
How are STS staged?
- STS are staged based on the TNM system by American joint committee on cancer.
- It involves size (T), lymph node spread (N), distant metastases (M) and histologic grade.
- Staging is mandatory since these factors have been shown to be independent in portraying the outcome of disease and treatment.
-
- T1: Tumour < 5 cm in dimension
- T2: Tumour > 5 cm in dimension
- A: Superficial tumour, b: Tumour Deep to subcutaneous tissues
- G1: low grade, G2: Intermediate grade, G3: high grade, G4: poorly differentiated on histology
- These parameters are evaluated and included in
- Stage I (A or B)
- Stage II (A or B)
- Stage III and Stage IV.
- Any lymph node involvement and Metastases in Stage IV.
What are the prognostic factors and survival for STS?
Prognosis for adult patients with STS are based on multiple independent factors such as age, tumour size, histologic grade, stage of tumour.
- Factors associated with poor prognosis are
- Age > 60 years
- Size > 5 cm
- High grade histology
- Prior intervention at non-oncology centers compromising local surgical removal margins.
- Survival is better when STS is located in extremities compared to poor in pelvic, retroperitoneal and head-neck areas.
- The 5-year currently observed survival rate for STS in extremities is as follows:
- Stage I: 90 %
- Stage II: 80 %
- Stage III: 55 %
- Stage IV: Extremely poor
How are Soft tissue sarcomas treated ?
- Optimal care for an individual with STS is provided by a multidisciplinary team consisting of musculoskeletal oncosurgeon, medical oncologist, radiologist, pathologist, radiation oncologist and social worker as in any sarcomas.
- Reports in scientific literature indicate STS treated at high volume centres have improved survival and functional outcomes due to multidisciplinary approach towards the cause.
The main treatment options include:
- Surgical removal with wide healthy tissue margins
- Radiation therapy
- Chemotherapy
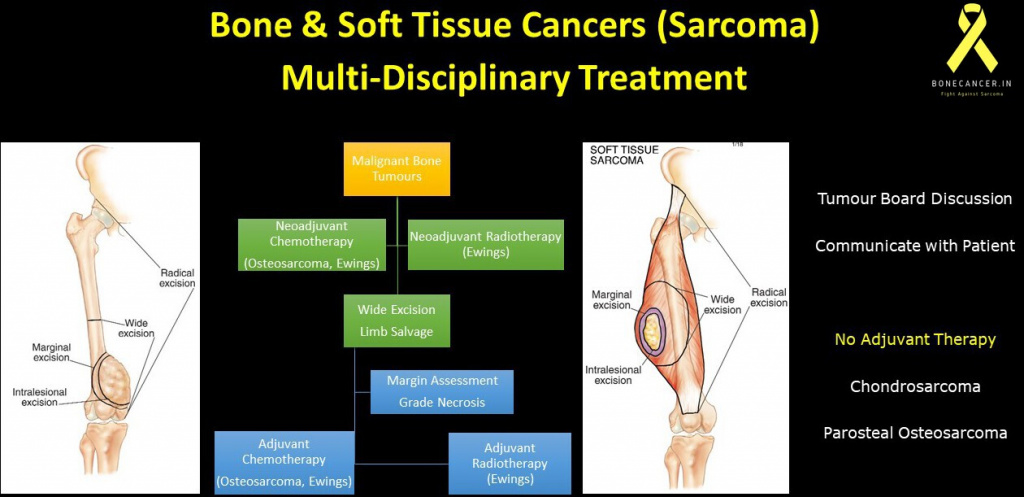
How are Soft tissue sarcoma treated surgically ?
- Stage I, Stage II and Stage III STS are all amenable for surgical removal including olio-metastatic stage IV STS.
- The aim of surgery is complete removal of sarcoma with “Wide healthy tissue margins” preserving the function of the involved extremity or body part.
- Often, removal of soft tissue sarcoma is followed by Micro-reconstructive procedures involving cover of defect by skin, muscle flaps and neurovascular reconstruction.
- Wide healthy margins may not be possible in all situations which are based on the aggressiveness of sarcoma and invasion into vital adjacent neurovascular structures.
- On these rare occasions, the critical neurovascular structures have to be sacrificed which may result in “Marginal removal” of sarcoma or radically even amputation of the involved limb.
- This extreme step is undertaken in consultation with both multi-disciplinary teams and the individual regarding locations of sarcoma, functional demand, need for adjuvant therapy and rehabilitation process.
What is the role of Radiotherapy in Soft Tissue Sarcoma?
- Radiotherapy can be provided to an individual with STS in two ways, Pre-operative and Post-operative.
- Post-operative radiotherapy complements adequate surgery as a standard treatment in intermediate or high grade, > 5 cm STS and also in marginal or contaminated surgical removals.
- Post-operative radiotherapy can be provided as brachytherapy and IMRT.
- It is started after a delay of 5-7 days after surgery to reduce wound healing problems (15-20 %).
- Post-operative radiation involves large doses and has risk of long term functional impairment.
- Brachytherapy: Involves placing tubes or catheter containing pellets in the tumour bed after surgical removal.
- This technique is commenced within a week of surgery and allows only the adequate and required amount of radiation to be delivered and spares the surrounding healthy tissues.
- Brachytherapy (even radical doses) has in scientific studies shown to improve outcome and reduce risk of local recurrence.
- External beam radiotherapy: This technique involves providing radiation therapy from outside the body.
- Newer techniques such as Intensity modulated radiation therapy (IMRT) are used often to deliver adequate amounts of radiation to specified areas.
- Certain centres also use Proton beam radiation to treat such instances.
- Pre-operative radiotherapy is provided to STS which are > 5cm, close proximity to major neurovascular structures and allows the surgeon to perform oncological safe and adequate surgical removal of the STS.
- Surgery is performed 4-6 weeks after radiotherapy.
- The major drawback is an increase in rate of wound complications after surgery (nearly 35 %) whereas has shown to slightly improve survival compared to postoperative radiotherapy.
What is the role of Chemotherapy in STS?
- The role of Chemotherapy in treating STS as neoadjuvant or adjuvant therapy is not clearly defined.
- It involves multiple drugs such as doxorubicin (Adriamycin), Ifosfamide and dacarbazine.
- Currently, chemotherapy is provided based on specific situations and as targeted therapies in few soft tissue sarcomas but no standard of treatment.
How is an individual with previous surgery for Unsuspected STS treated?
- There are instances when an individual undergoes a surgical intervention attempting cure of Lump or Mass without any suspicion of STS and approaches an oncology center for another opinion.
- Post-surgery, the histopathological diagnosis indicates a sarcomatous lesion.
- This scenario is considered to be of Inadequate surgery for a suspected STS and investigated step-wise similar to a newly diagnosed STS.
- Subsequently, in an ideal condition this STS is treated surgically with wide healthy tissue margins followed by adjuvant therapy.
- This systematic approach of repeat surgery to obtain oncological clearance has shown to improve the outcome and survival compared to just providing adjuvant therapy in presence of inadequate surgery.
How is an individual with STS monitored after treatment?
- An individual who has completed treatment for STS and shows no active disease is required to visit the treating musculoskeletal Oncosurgeon regularly to monitor for any local recurrence or metastases.
- The risk assessment based on tumour grade, size, and site are beneficial in choosing the routine follow up customized to each individual.
- The visit intervals are every 3 months for the first 2 years followed by every 6 months for the next 2 years and every year subsequently.
- Every visit involves physical examination and CT Thorax followed by FDG PET CT annually.
It is not absolutely necessary to perform MRI of the involved region at every visit or interval for every individual except in situations of high risk such as large volume disease, Stage III or IV at presentation, positive physical examination during follow up or complicated case scenarios.
How is local recurrence and metastatic disease in STS treated?
- Local recurrence is identified by physical examination followed by UGS or MRI scan.
- Surgical removal is advised based on the morphology, location and extent of local recurrence.
- Metastases appear in 20 % of individuals with STS.
- It is common in lungs and rare in other sites.
- Metastases in both small numbers and sizes are advised for metastasectomy (removal) based on the general condition of the individual.
-
- This aggressive approach has shown to improve the outcome and survival in treatment of STS.
- Factors associated with prolonged survival are complete clearance of lung metastases and greater than 1 year disease free interval from primary surgery.
- Hence, any oncological progression in STS does not always result in poor prognosis.